PAM-XIAMEN può fornire substrato monocristallino AlN, fare riferimento a specifiche aggiuntivehttps://www.powerwaywafer.com/aln-substrate.html.
I principali droganti candidati per l'AlN di tipo n sono l'ossigeno (O) e il silicio (Si), mentre per l'AlN di tipo p sono il magnesio (Mg) e il berillio (Be). Finora, il tasso di successo del doping con Mg e O è stato molto basso. Tuttavia, O è un'importante impurità di compensazione sia per Mg che per Be, poiché sia i droganti che l'Al stesso tendono ad assorbire ossigeno. Per comprendere il drogaggio dell'AlN, è necessario comprendere il ruolo dei posti vacanti di azoto (VN) e di alluminio (VAL) e dei relativi complessi. L'attuale comprensione della chimica dei difetti e delle impurità, nonché dei metodi di drogaggio non in equilibrio, sarà considerata un approccio promettente per superare gli attuali limiti tecnologici.
1.DonorDfunzionamento di AlN:Silicona
Per AlN di tipo n, il silicio è il donatore teorico ottimale di cationi perché il suo raggio atomico è molto vicino a quello di Al, come mostrato in Fig. 1 (Al è 118 pm, Si è 111 pm). Sebbene il silicio sia un donatore superficiale in GaN con un'energia di attivazione ED di circa 17 meV, in AlGaN, con un aumento del contenuto di Al, la sua ED aumenta da 24 meV in Al0,85Ga0,15N a 211 meV in Al0,96Ga0,04N. Il silicio, come impurità sostitutiva in AlN, provoca una contrazione teorica del 6% nel legame N del piano basale più vicino. Ciò indica una diminuzione della posizione del reticolo di silicio, più vicino ai tre legami basali, durante la quale i legami Si-N dell'asse c vengono allungati. Quando il silicio cattura un elettrone secondario e subisce un riarrangiamento geometrico, si forma un centro DX, che include una contrazione del 2% dei legami Si-N in tre posizioni basali e una rottura del legame Si-N sull'asse c, come mostrato in Fig. 1, portando ad una transizione da stati superficiali a stati profondi. Quando il legame Si-N dell'asse c sotto stress si rompe, verrà generato uno stato profondo compensato. Nonostante l'esistenza di tali difetti di compensazione, il drogaggio di tipo n dell'AlN attraverso l'impianto ionico nella regione vicina alla superficie ha mostrato risultati promettenti. Tuttavia, nel drogaggio di tipo n di film sottili MOCVD AlN non compensati, solo una concentrazione di elettroni riproducibile fino a 1015centimetro-3viene visualizzato, applicabile solo alle regioni con campo di deriva elevato nel dispositivo.
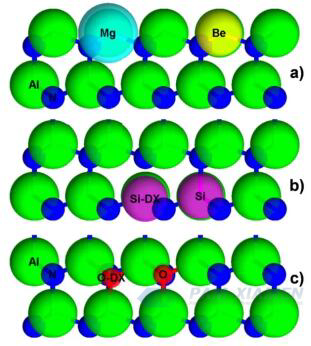
Fig. 1 Le posizioni e le dimensioni relative (in proporzione) di (a) accettori di Mg e Be in AlN, (b) impurità di Si riorganizzate da donatori di Si e DX e (c) posizioni e dimensioni relative di difetti di ossigeno sostituiti con ossigeno e Riarrangiamenti DX (in proporzione)
Dato che una tecnologia dimostra la fattibilità del doping mentre un’altra tecnologia no, il silicio stesso non può essere il problema. L'ipotesi che il non equilibrio e l'MBE a bassa temperatura contribuiscano al drogaggio di tipo n dell'AlN si basa sui seguenti due punti:
1) quando si utilizza la bassa temperatura, si ottiene l'espansione termica minima, pertanto è desiderabile l'allungamento minimo aggiuntivo del legame Si-N sull'asse c;
2) Riducendo al minimo le condizioni di arricchimento del metallo per i complessi Al di posti vacanti Si, questi complessi mostrano un comportamento simile ai complessi di Al posti vacanti O, portando all'autocompensazione del doping del donatore a livelli elevati di drogaggio di Si.
Si ipotizza che questa autocompensazione del Si sia dovuta all'ammorbidimento del reticolo causato da VAl, che rende più facile la rottura del legame Si-N dell'asse c e quindi la formazione di un centro Si-DX compensato. Similmente a O, per la neutralità di carica, va considerato anche il complesso VAL-XSiAl, dove X è un numero intero. Pertanto, dichiarare un MBE non in equilibrio, a bassa temperatura e arricchito con metalli è molto utile per il drogaggio di tipo n di AlN.
2. AccettoreDfunzionamento di AlN:Mg, Essere, E C
Per AlN di tipo p, gli accettori, come il carbonio sostituito con anioni (C) e Be e Mg sostituiti con cationi, sono considerati fattibili. Quando si avvicinano al limite di diluizione, le energie degli accettori isolati fanno sì che questi accettori vengano considerati come recettori di livello profondo, con energie di attivazione degli accettori a valore singolo teoricamente riportate per Mg e Be che vanno rispettivamente da 510 a 630 meV e da 220 a 340 meV. Se non vi è la capacità di formare bande di impurità per ridurre queste grandi energie di attivazione, non è prevista una concentrazione significativa dei pori.
Tuttavia, come mostrato dalla sovrapposizione orbitale di Bohr ad alte concentrazioni di drogaggio in GaN, è possibile che l'energia di attivazione effettiva del Mg diminuisca da circa 210 meV a circa 50 meV, risultando in una concentrazione dei pori di 1 x 1020centimetro-3. Ciò si è dimostrato efficace anche per AlGaN con un massimo di circa il 60% di Al e sono stati dimostrati i diodi tunnel.
A parte una concentrazione di pori di circa 1010 cm-3, Mg non è stato utilizzato con successo per drogare AlN. I recenti risultati dell'utilizzo del Be per ottenere una concentrazione dei pori a temperatura ambiente di circa 3 x 1018centimetro-3(riportato qui come circa 4,4 x 1018centimetro-3) e un'energia di attivazione effettiva di circa 37 meV indicano che è possibile ottenere una riduzione dell'energia di attivazione.
Le costanti dielettriche relative del nitruro di gallio (GaN), del nitruro di alluminio (AlN) e del nitruro di indio (InN) sono rispettivamente 8,9, 8,5 e 15,3. Le masse effettive riportate dei buchi pesanti sono rispettivamente 0,8mo, 3,53mo e 1,63mo. Ciò significa che le concentrazioni critiche di Mott formate dalle bande accettrici di GaN, AlN e InN sono circa 4×1019cm-3, 4×1021cm-3e 6,5×1019centimetro-3, rispettivamente. Questa approssimazione semplificata è in buon accordo con i risultati del drogaggio degli esperimenti di epitassia modulata con metallo (MME) per GaN, dove le concentrazioni di fori nell'intervallo di 1019centimetro-3possono essere raggiunti regolarmente. Allo stesso modo, queste previsioni indicano che AlN e InN saranno più difficili da subire un drogaggio di tipo p degenerato, poiché ciascuno richiede una concentrazione di drogaggio più elevata per formare bande di impurità.
Per AlN, inizialmente può essere considerato impossibile raggiungere l'elevata concentrazione di drogaggio richiesta per formare bande di impurità, il che è contrario agli attuali risultati sperimentali. Tuttavia, è necessario considerare la struttura unica della banda di valenza di AlN, dove la banda di separazione è effettivamente più alta della banda delle lacune e della banda delle lacune leggere (vedere Fig. 2) e presenta un'elevata anisotropia. A causa della mancanza di elettroni dell'orbitale d, il segno dell'energia di scissione del campo cristallino è opposto a quello del GaN. La banda di scissione presenta una curvatura maggiore e si trova su una gamma più ampia di bande di fori pesanti e leggeri in termini di energia. La Fig. 2 mostra il calcolo della teoria del funzionale della densità (DFT) considerando questo effetto. Le masse efficaci calcolate dalla teoria del funzionale della densità (DFT) sono 3,66mo per la componente parallela e 0,24mo per la componente verticale, risultando in concentrazioni critiche di Mott di 4,3×1021centimetro-3e 1,2×1018centimetro-3, rispettivamente. I limiti superiore e inferiore della concentrazione critica di Mott sono forniti per enfatizzare che, considerando l'unicità della struttura delle bande di AlN, è possibile prevedere la formazione di bande di impurità in AlN, che possono portare a una maggiore mobilità delle lacune rispetto a GaN, portando speranza per dispositivi AlN a canale p e bipolari.
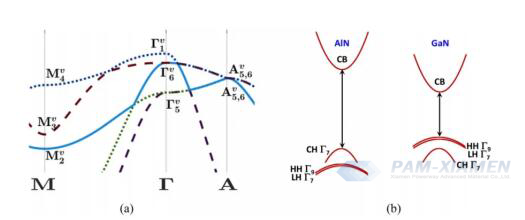
Fig. 2 (a) Dispersione elettronica funzionale densità della traiettoria lineare della banda di valenza AlN non sollecitata (senza spin) della wurtzite lungo i punti di simmetria M, Γ e A all'interno della zona di Brillouin. Al centro della regione, gli stati delle bande separate ΓV1 sono degenerazione di singoletto, mentre gli stati dei buchi pesanti e leggeri ΓV 6 sono doppiamente degenerati. (b) Dispersione elettronica qualitativa della wurtzite AlN e GaN non sollecitata lungo la direzione k⊥. A differenza del GaN, la banda di valenza più alta nell'AlN è la banda di dissociazione, che presenta una massa effettiva più piccola. L'introduzione dell'interazione dell'orbita di spin allevia la degenerazione delle bande di buchi pesanti e leggeri nel centro della regione, determinando l'emergere di un singolo stato degenerato ΓV; 17, ΓV9 e ΓV;27.

Per ulteriori informazioni potete contattarci via e-mail all'indirizzovictorchan@powerwaywafer.com e powerwaymaterial@gmail.com.