PAM-XIAMEN có thể cung cấp chất nền đơn tinh thể AlN, thông số kỹ thuật bổ sung vui lòng tham khảohttps://www.powerwaywafer.com/aln-substrate.html.
Các chất dẫn xuất AlN loại n chính là oxy (O) và silicon (Si), trong khi đối với AlN loại p, chúng là magie (Mg) và berili (Be). Cho đến nay, tỷ lệ thành công của việc sử dụng doping Mg và O rất thấp. Tuy nhiên, O là tạp chất bù quan trọng cho cả Mg và Be, vì cả chất tạp chất và bản thân Al đều có xu hướng hấp thụ oxy. Để hiểu được sự pha tạp của AlN, cần phải hiểu vai trò của các khuyết nitơ (VN) và các khuyết nhôm (VAl) và các phức chất liên quan của chúng. Sự hiểu biết hiện tại về khuyết tật và hóa học tạp chất, cũng như các phương pháp doping không cân bằng, sẽ được coi là một cách tiếp cận đầy hứa hẹn để khắc phục những hạn chế về công nghệ hiện tại.
1. Dtrên hoặcDhoạt động của AlN:Sbiểu tượng
Đối với AlN loại n, silicon là chất cho cation lý thuyết tối ưu vì bán kính nguyên tử của nó rất gần với bán kính nguyên tử của Al, như trong Hình 1 (Al là 118 chiều, Si là 111 chiều). Mặc dù silicon là chất cho nhỏ trong GaN với năng lượng kích hoạt ED xấp xỉ 17 meV, nhưng trong AlGaN, khi hàm lượng Al tăng lên, ED của nó tăng từ 24 meV trong Al0.85Ga0.15N lên 211 meV trong Al0.96Ga0.04N. Silicon, như một tạp chất thay thế trong AlN, gây ra sự co lại về mặt lý thuyết là 6% trong liên kết N mặt phẳng cơ bản lân cận gần nhất. Điều này cho thấy sự giảm vị trí của mạng silicon, gần hơn với ba liên kết cơ bản, trong đó các liên kết trục Si-N bị kéo căng. Khi silicon bắt giữ một electron thứ cấp và trải qua quá trình sắp xếp lại hình học, một tâm DX được hình thành, bao gồm sự co lại 2% của các liên kết Si-N ở ba vị trí cơ bản và sự đứt gãy liên kết Si-N trục c, như trong Hình 1, dẫn tới sự chuyển đổi từ trạng thái nông sang trạng thái sâu. Khi liên kết Si-N trục c bị đứt do ứng suất, trạng thái sâu được bù sẽ được tạo ra. Bất chấp sự tồn tại của những khiếm khuyết bù như vậy, việc pha tạp AlN loại n thông qua cấy ion ở vùng gần bề mặt đã cho thấy kết quả đầy hứa hẹn. Tuy nhiên, trong pha tạp loại n của màng mỏng MOCVD AlN không bù, chỉ có nồng độ electron tái tạo lên tới 1015cm-3được hiển thị, chỉ áp dụng cho các vùng trường có độ lệch cao trong thiết bị.
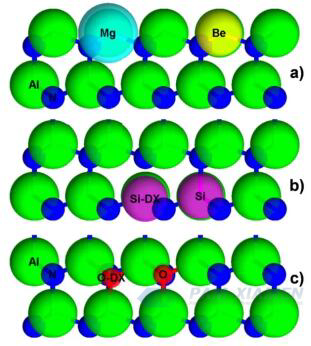
Hình 1. Vị trí và kích thước tương đối (theo tỷ lệ) của (a) chất nhận Mg và Be trong AlN, (b) tạp chất Si được sắp xếp lại bởi các chất cho Si và DX, và (c) vị trí và kích thước tương đối của các khuyết tật oxy được thay thế cho oxy và Sắp xếp lại DX (theo tỷ lệ)
Cho rằng một công nghệ chứng minh tính khả thi của doping trong khi công nghệ khác thì không, bản thân silicon không thể là vấn đề. Giả thuyết cho rằng MBE không cân bằng và nhiệt độ thấp góp phần tạo ra pha tạp loại n của AlN dựa trên hai điểm sau:
1) khi sử dụng nhiệt độ thấp, đạt được độ giãn nở nhiệt tối thiểu, do đó mong muốn độ giãn dài liên kết Si-N bổ sung tối thiểu trên trục c là mong muốn;
2) Giảm thiểu các điều kiện làm giàu kim loại cho các phức hợp Al trống Si, các phức này thể hiện hành vi tương tự như các phức hợp Al trống O, dẫn đến việc tự bù pha tạp chất cho ở mức pha tạp Si cao.
Người ta suy đoán rằng sự tự bù Si này là do sự làm mềm mạng do VAl gây ra, khiến cho việc phá vỡ liên kết Si-N trục c dễ dàng hơn và do đó hình thành trung tâm Si-DX được bù. Tương tự như O, do tính trung hòa điện tích nên phức VAl-XSiAl cũng phải được xét đến, trong đó X là số nguyên. Do đó, khẳng định MBE không cân bằng, nhiệt độ thấp và giàu kim loại là rất có giá trị đối với pha tạp loại n của AlN.
2. Người chấp nhậnDhoạt động của AlN:Mg, Hãy, Và C
Đối với AlN loại p, các chất nhận, như carbon thay thế anion (C) và Be và Mg được thay thế cation được coi là khả thi. Khi đạt đến giới hạn pha loãng, năng lượng của chất nhận bị cô lập dẫn đến những chất nhận này được coi là thụ thể ở mức độ sâu, với năng lượng kích hoạt chất nhận có giá trị duy nhất được báo cáo về mặt lý thuyết đối với Mg và Be lần lượt nằm trong khoảng từ 510 đến 630 meV và 220 đến 340 meV. Nếu không có khả năng hình thành các dải tạp chất để giảm năng lượng kích hoạt lớn này thì nồng độ lỗ rỗng đáng kể sẽ không được mong đợi.
Tuy nhiên, như thể hiện qua sự chồng lấp quỹ đạo Bohr ở nồng độ pha tạp cao trong GaN, năng lượng hoạt hóa hiệu quả của Mg có thể giảm từ khoảng 210 meV xuống còn khoảng 50 meV, dẫn đến nồng độ lỗ rỗng là 1 x 1020cm-3. Điều này cũng đã được chứng minh là có hiệu quả đối với AlGaN với hàm lượng Al lên tới khoảng 60% và điốt đường hầm đã được chứng minh.
Ngoài nồng độ lỗ rỗng khoảng 1010 cm-3, Mg chưa được sử dụng thành công để pha tạp AlN. Kết quả gần đây của việc sử dụng Be để thu được nồng độ lỗ rỗng ở nhiệt độ phòng khoảng 3 x 1018cm-3(được báo cáo ở đây là khoảng 4,4 x 1018cm-3) và năng lượng kích hoạt hiệu dụng khoảng 37 meV cho thấy có thể đạt được mức giảm năng lượng kích hoạt.
Hằng số điện môi tương đối của gali nitrit (GaN), nhôm nitrit (AlN) và indi nitrit (InN) lần lượt là 8,9, 8,5 và 15,3. Khối lượng hiệu dụng được báo cáo của lỗ nặng lần lượt là 0,8mo, 3,53mo và 1,63mo. Điều này có nghĩa là nồng độ tới hạn Mott được hình thành bởi các dải chấp nhận của GaN, AlN và InN là khoảng 4×1019cm-3, 4×1021cm-3và 6,5×1019cm-3, tương ứng. Phép tính gần đúng đơn giản hóa này phù hợp tốt với kết quả pha tạp của các thí nghiệm epit Wax điều chế kim loại (MME) đối với GaN, trong đó nồng độ lỗ trống trong khoảng 1019cm-3có thể đạt được thường xuyên. Tương tự, những dự đoán này chỉ ra rằng AlN và InN sẽ khó trải qua quá trình pha tạp loại p thoái hóa hơn, mỗi loại đòi hỏi nồng độ pha tạp cao hơn để hình thành các dải tạp chất.
Đối với AlN, ban đầu có thể được coi là không thể đạt được nồng độ pha tạp cao cần thiết để hình thành các dải tạp chất, điều này trái ngược với kết quả thí nghiệm hiện tại. Tuy nhiên, cần phải xem xét cấu trúc dải hóa trị duy nhất của AlN, trong đó dải tách ra thực sự cao hơn dải lỗ và dải lỗ nhẹ (xem Hình 2) và có tính dị hướng cao. Do thiếu electron quỹ đạo d nên dấu hiệu năng lượng phân tách trường tinh thể trái ngược với dấu hiệu của GaN. Dải tách ra thể hiện độ cong lớn hơn và nằm trên dải năng lượng rộng hơn của các dải lỗ nặng và nhẹ. Hình 2 cho thấy phép tính lý thuyết hàm mật độ (DFT) có xét đến hiệu ứng này. Khối lượng hiệu dụng được tính theo lý thuyết hàm mật độ (DFT) là 3,66mo đối với thành phần song song và 0,24mo đối với thành phần thẳng đứng, dẫn đến nồng độ tới hạn Mott là 4,3×1021cm-3và 1,2×1018cm-3, tương ứng. Giới hạn trên và giới hạn dưới của nồng độ tới hạn Mott được đưa ra để nhấn mạnh rằng khi xem xét tính duy nhất của cấu trúc dải AlN, có thể dự đoán sự hình thành các dải tạp chất trong AlN, có thể dẫn đến độ linh động lỗ trống cao hơn GaN, mang lại hy vọng. cho các thiết bị AlN kênh p và lưỡng cực.
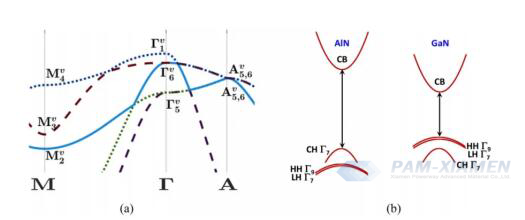
Hình 2 (a) Sự phân tán electron chức năng mật độ của quỹ đạo tuyến tính của dải hóa trị AlN không bị nén (không có spin) của wurtzite dọc theo các điểm đối xứng M, Γ và A trong vùng Brillouin. Ở trung tâm của vùng, các trạng thái dải tách rời ΓV1 là trạng thái thoái hóa đơn, trong khi trạng thái lỗ nặng và lỗ nhẹ ΓV 6 là suy biến kép. (b) Sự phân tán electron định tính của wurtzite AlN và GaN không chịu ứng suất dọc theo hướng k⊥. Không giống như GaN, dải hóa trị cao nhất trong AlN là dải phân ly, thể hiện khối lượng hiệu dụng nhỏ hơn. Sự ra đời của tương tác quỹ đạo quay làm giảm sự suy biến của các dải lỗ nặng và nhẹ ở trung tâm vùng, dẫn đến sự xuất hiện của trạng thái suy biến đơn ΓV; 17, ΓV9 và Γ V;27.

Để biết thêm thông tin, xin vui lòng liên hệ với chúng tôi email tạivictorchan@powerwaywafer.com và powerwaymaterial@gmail.com.